从maizegdb上下载的序列无法比对到ncbi上去
包括编码链和非编码连的比对,而NCBI中提交的序列都是编码链的序列你在用DNAMAN的多序列比对时,在弹出的对话框中可能没有选择\\“尝试使用双链”吧,你测序得到的目的片段可能是非编码链(即你目的基因的编码链的互补序列)我要做一个系统树,序列是从NCBI上粘贴下来的,但是word和txt格式无法Clustal比对,有高人能指点一下么?
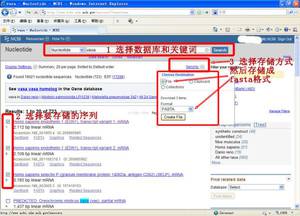
从NCBI上可以直接下载各种格式的序列文件,clustal是可以读取fasta格式的。具体看看图吧。
图示搜索核酸序列之后,选择自己想要的序列然后下载到本地磁盘。
序列下载下来就可以用clustal打开了。
另外,最好不要把fasta序列文件放到桌面,貌似有些版本的clustal不能装入放在桌面的序列文件。
NCBI的使用和进化树构建求助
因此NCBI 的分类学数据库不是一个系统发育或分类学的“专家数据库”(Wheeler et al., 2000)。 获取序列所对应的分类学信息有两种方法。 一种方法,从NCBI 网站下载gi与taxid 对应表,在Taxonomy 数据库的FTP 地址下载。这个目录下有多个压缩文件,其中针对Windows 操作系统的两个针对蛋白质序列和核苷酸序列的压缩文件分别是gi_taxid_prot.dmp.gz 和gi_taxid_nucl.dmp.gz 文件。这两个文件都只有两列,左边为gi 号,右边为Taxid。由于这些文件非常大,因此用浏览器来打开这些文件几乎是不可能的。随着时间的推移,这两个文件如何构建系统发育树
无论是基于距离的系统发生树重建方法,还是基于特征的系统发生树重建方法,都不能保证一定能够得到一棵描述比对序列进化历史的真实的树。
一般地,对于某个数据集,如果用一种方法能推断出正确的系统发生关系,则用其它流行的方法也能得到较好的结果。但是,如果模拟数据集中序列的变化很大,或不同的分支变化速率不同,则没有一种方法是十分可靠的。
扩展资料:
系统树是一种分支图(英文cladogram)。在树中,每个节点代表其各分支的近共同祖先,而节点间的线段长度对应演化距离(如估计的演化时间)。
系统发生树有时也称系统树图,它是由一系列节点和分支组成的。其中每一个节点代表一个分类单元,分支末端的节点对应 一个基因或者生物体。与外部节点对应,内部节点代表一个推断出的共同祖先。系统发生树结构的基本信息在计算机程序中常常用一组嵌套的圆括号表示,成为newick格式。
参考资料来源:百度百科-系统发生树